Introductions
HBIcloud is a comprehensive online platform designed to facilitate multi-omics data analysis by integrating the various tools of genomics, transcriptomics, proteomics, metabolomics, phenomics and multi-omics integration. Developed to address the growing complexity and volume, HBIcloud provides researchers with a powerful and user-friendly resource to perform complex analyses without the use of extensive programming skills. With more than 100 tools, the platform offers standardized workflows, extensive parameter options, and rich documentation to meet the diverse needs of the scientific community. The research behind HBIcloud aims to create a centralized, user-friendly platform to streamline the analysis process, allowing researchers to focus on scientific discoveries rather than technical challenges. By integrating various tools and providing extensive support and documentation, HBIcloud addresses the critical need for standardized, repeatable workflows in multi-omics research.
Input files & output files
The formats of csv , xlsx, xlm, xlsm , txt , bam, sam, fasta, fastq, vcf, newick , etc. The .csv file stores table data (numbers and text) in plain text form. It is recommended to use WORDPAD or Notepad to open it, or save it as a new file and then open it with EXCEL.
The input file needs to be uploaded within 500M
The output file is image, which includes scalar images such as png and jpg, and vector images such as svg.
1.Operation instruction
To meet the demands for correlational analysis and visualization of massive multidimensional omics data, we have initiated the HBIcloud Bioinformatics Cloud Platform. This comprehensive bioinformatics tool is designed to provide researchers with convenient and efficient solutions for data processing and analysis. HBIcloud offers a range of powerful tools, including genome sequence analysis, transcriptome analysis, protein sequence alignment, evolutionary analysis, statistical analysis, and more, enabling in-depth exploration of species structure and function. Furthermore, HBIcloud provides a wealth of data visualization tools, such as bar charts, line graphs, heatmaps, etc., assisting users in intuitively presenting and analyzing data. Integrated within HBIcloud are molecular breeding tools, facilitating genetic and breeding research. We are committed to continuously updating and optimizing the platform's tools and features to ensure users always have access to the latest bioinformatics solutions. User data on the HBIcloud platform is rigorously protected through multiple security measures to ensure data integrity and privacy. Whether in the field of biology or medicine, the HBIcloud Bioinformatics Cloud Platform offers comprehensive support for data processing and analysis.
1.1 Home page
① The navigation bar contains 100+ tools of eight omics. Click the tool name to jump to the corresponding tool interface
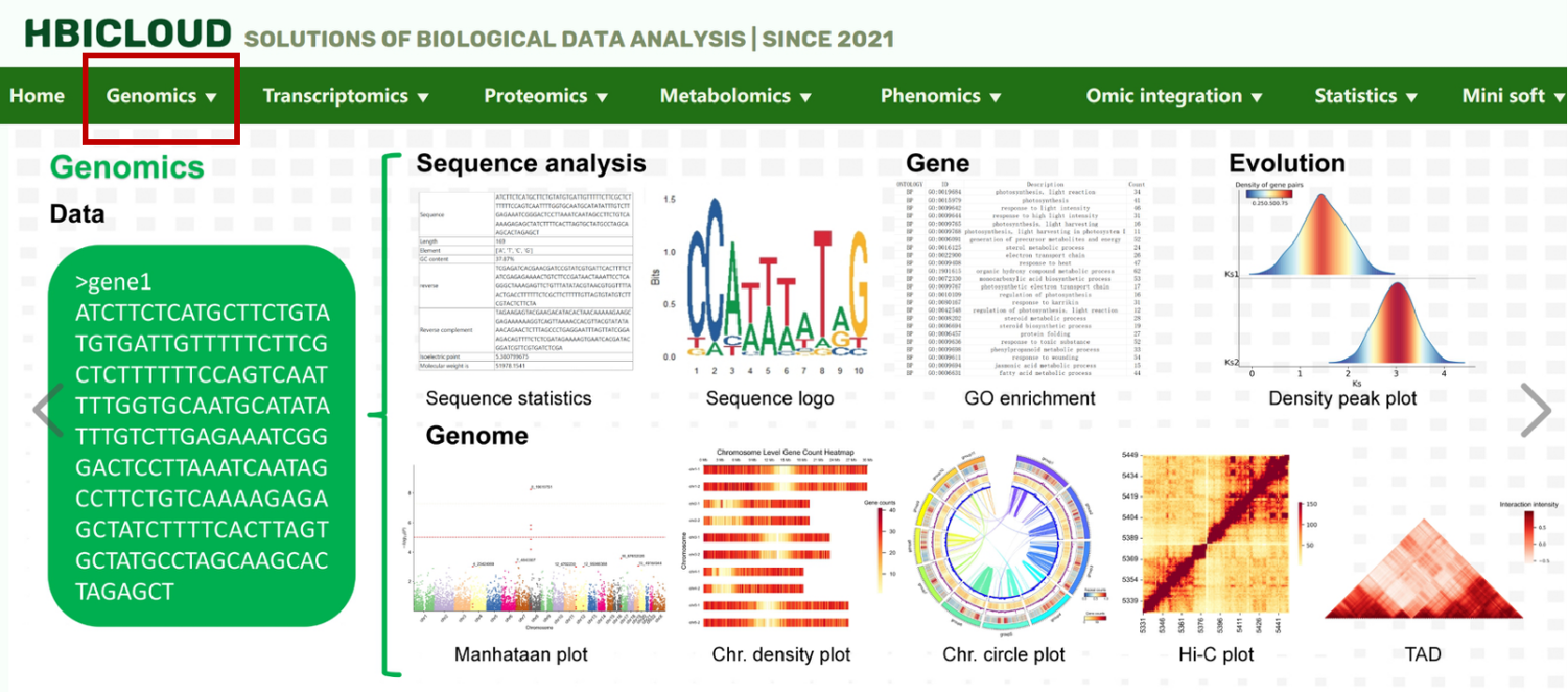
② In the middle area, there are instructions, FAQ, related meetings, introduction of our team, page jump links to other omics links, and pie charts of tools statistics of this platform, as well as six special highlights

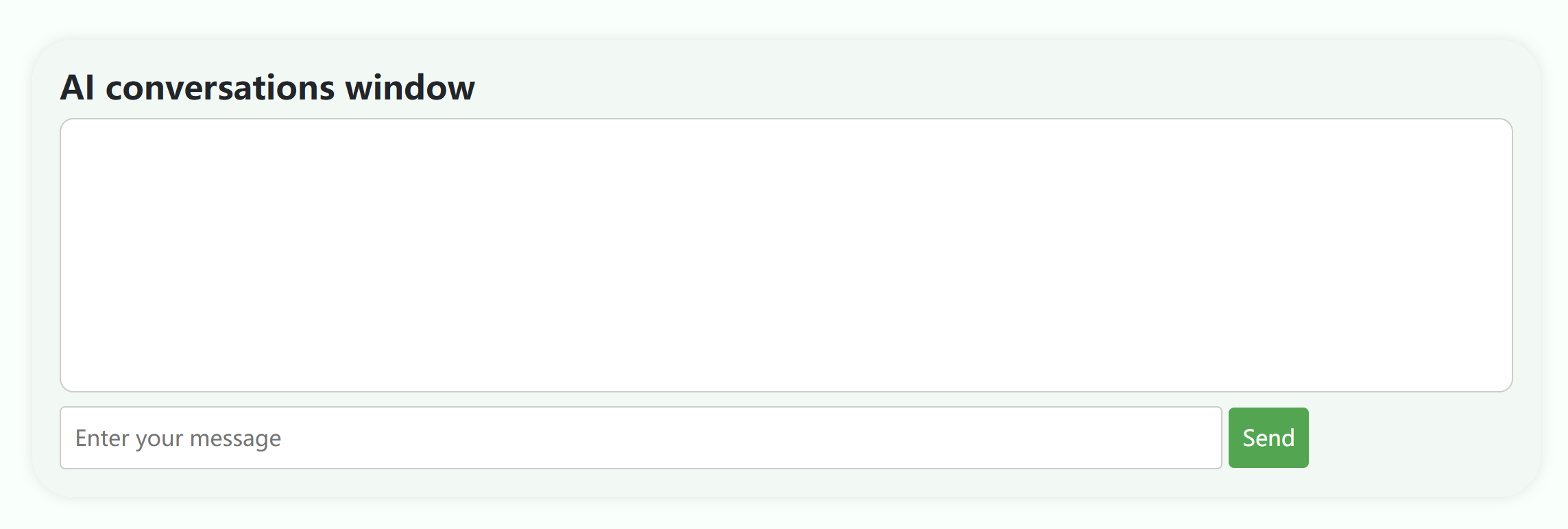
③ The following is the AI question and answer window. By entering a question at the bottom, the AI will answer the user's question.
④ The bottom area introduces the update of the platform, the information of the platform designer, the unit of the platform user, and the information of the platform visitor.

1.2 Tools page
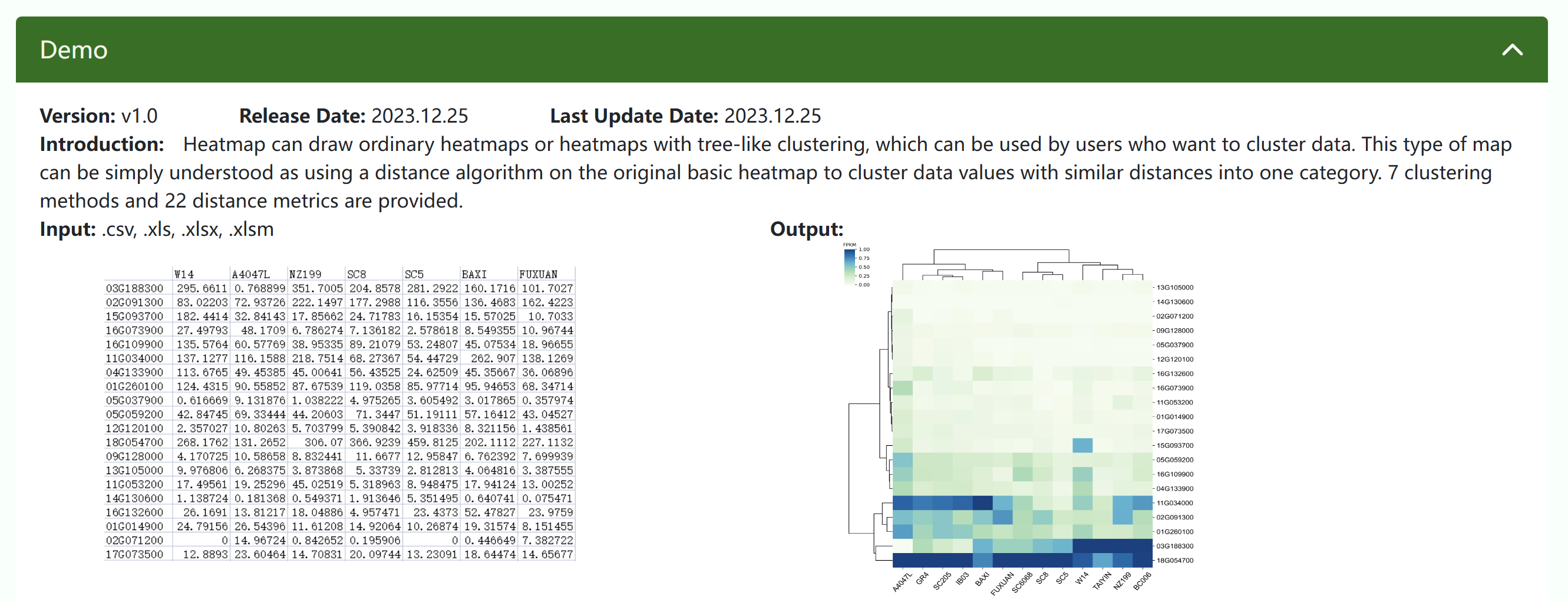
① Demo describes the version, development time, update time, tool introduction, input file format, input file preview, and output file preview of the tool.
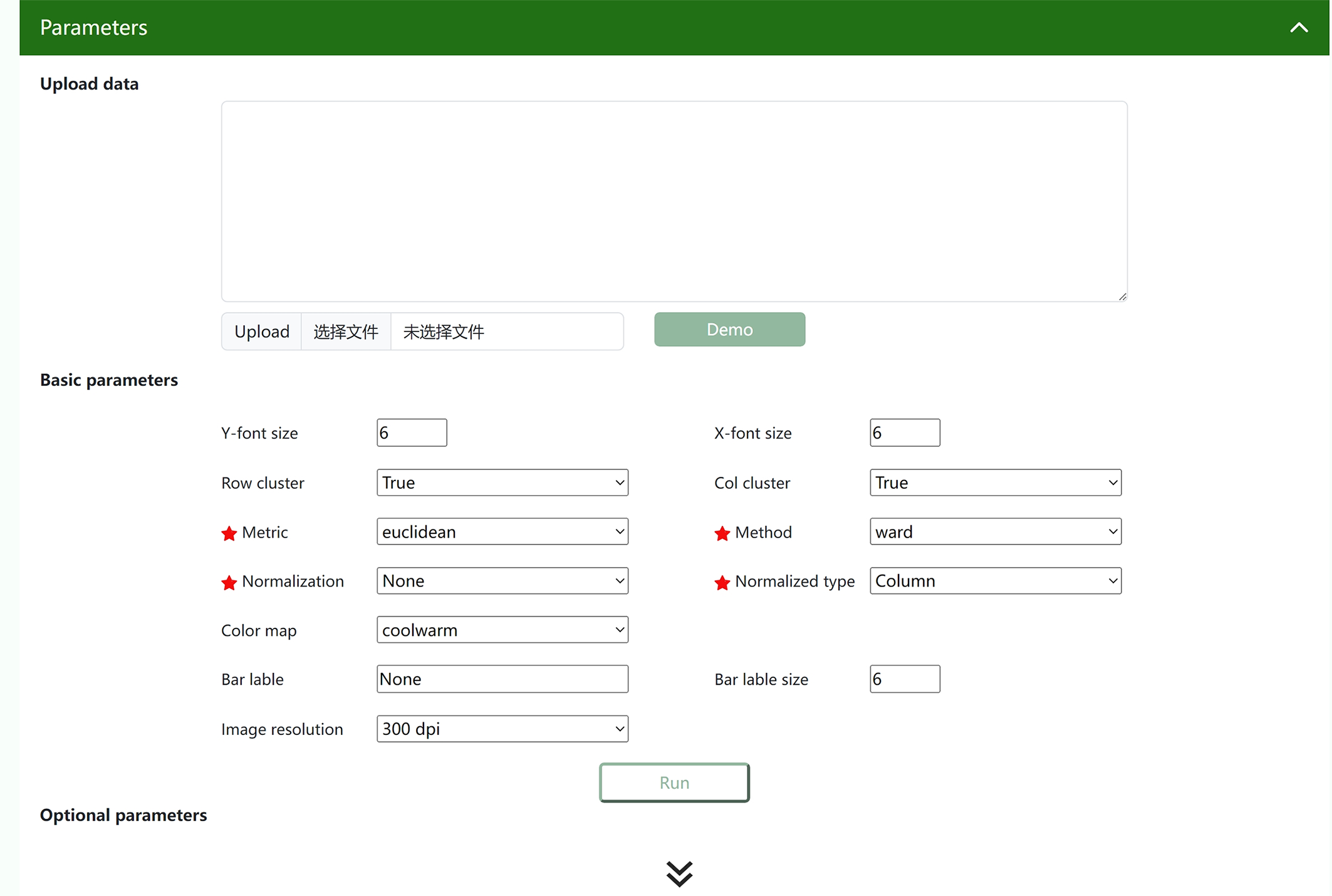
② You can set basic parameters or drop down advanced parameters to adjust them. After adjusting the parameters, click "Run".
2.1 Sequence analysis
Sequence statistics
It is used to perform statistical analysis on biological sequences and provide information on basic characteristics such as sequence length, base/amino acid composition, GC content, etc. These statistics can help researchers identify the basic properties of sequences and provide a basis for further functional prediction and comparative analysis.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Online time: 2023.12.25
Update time: 2023.12.25
Sequence editing
Involving the modification and editing of biological sequences, including insertion, deletion, and substitution of bases or amino acids, it enables researchers to precisely modify target sequences to study gene function, create model organisms, or develop gene therapy methods .
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- The "Element" parameter inputs the element that needs to be replaced
- "Replace element" parameter input replacement element
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 2023.12.25
Update time: 2023.12.25
Sequence editing
Sequence extraction tool isolates specific DNA, RNA, or protein sequences from larger datasets, streamlining the analysis in genomics and proteomics. It aids in identifying key biological patterns or variations for downstream studies.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- The "Sequence ID" parameter inputs the gene name
- The "Start" parameter is used to input the sequence position where extraction begins
- The "End" parameter input indicates the position of the extracted sequence
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 2024.8.12
Update time: 2024.8.12
Multiple sequence extraction
Multiple sequence extraction tool isolates specific DNA, RNA, or protein sequences from larger datasets, streamlining the analysis in genomics and proteomics. It aids in identifying key biological patterns or variations for downstream studies.
Detailed instructions:
1. First, click the "Upload" button in the "Upload data" area to enter the sequence file and the location file that needs to be extracted.
2. After completing all configurations, click the "Run" button to start running.3. The output results are displayed in the Result area.
Online time: 2024.8.12
Update time: 2024.8.12
Sequence logo
A graphical representation method for visualizing the results of multiple sequence alignments, showing the conservation and variability of sequences at specific positions. By illustrating the frequency of different bases or amino acids at each position, researchers can quickly identify conserved regions and functionally important sites, and then infer their biological functions.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- "Data type" parameter input data type
- The "Number of columns" parameter sets the number of columns, which is only valid when facet='wrap'
- The "Type" parameter sets the sequence type and can be one of "auto", "aa", "dna", "rna" or "other"
- The "Method" parameter sets two sequence marker generation methods: bits and probability
- "Lable size" parameter sets the font size
- "Stack width" parameter sets the letter stack width between 0 and 1
- "Color scheme" parameter applies the color scheme to the sequence logo.
- "Facet" parameter input facet type
- label name parameters for the X-axis and Y-axis
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 2023.12.25
Update time: 2023.12.25
Domain plot
Used to display the distribution of domains in a protein or gene sequence. By illustrating different domains and their locations and lengths in the sequence, researchers can understand the functional modules of proteins, analyze their structure and function relationships, and predict the functions of unknown proteins.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- The "Feature" parameter selects whether a feature file needs to be input
- "Feature file" parameter input feature file
- "Mark shape" parameter selects the shape of the mark
- "Aligning" parameter is aligned, input alignment gene
- The "Element lable" parameter sets whether to display element labels
- Set the width and height parameters
- The "Color map" parameter contains 166 types, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Set the font size and label size of the X and Y axes
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 2024.6.13
Update time: 2024.6.13
2.2 Gene
Gene cluster prediction
Used to predict gene clusters, which are regions of the genome where functionally related genes are clustered. This helps study gene co-expression, metabolic pathways, and biosynthetic pathways.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024. 5. 8
Updated: 2024. 5. 8
TSS extract
Extracting transcription start sites helps study gene expression regulation, transcription mechanism and gene promoter activity.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024. 5. 8
Updated: 2024. 5. 8
Promoter extract
Extract gene promoter sequences to help study the regulatory elements in the promoter region and the regulatory mechanism of gene expression.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Online time: 2024.6.13
Update time: 2024.6.13
KEGG
Use the KEGG database to perform functional annotation of genes and proteins to help understand metabolic pathways and molecular interaction networks.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- "Qvalue" parameter inputs the q value threshold
- "Pvalue" parameter input p-value threshold
- "Padjust method" parameter input multiple testing correction method
- "Ont" parameter inputs enrichment pathway type
- The "Readable" parameter sets the output format
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 2024.6.13
Update time: 2024.6.13
GO
Gene Ontology is used to annotate genes functionally, with classifications including biological processes, cellular components, and molecular functions, to help understand the biological functions of gene products.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- "Qvalue" parameter inputs the q value threshold
- "Pvalue" parameter input p-value threshold
- "Padjust method" parameter input multiple testing correction method
- The "database" parameter sets whether the species is in the KEGG database
- "Annotation file" parameter If "database" is selected, you need to enter the species annotation file
- Enter the abbreviation of the species in the KEGG database in the "Organism" parameter. For details, please refer to the link https://www.genome.jp/kegg/tables/br08606.html
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 2024.6.13
Update time: 2024.6.13
2.3 Genome
Hi-C plot
Hi-C is an experimental technique for studying the three-dimensional conformation of the whole genome and analyzing the interaction of chromatin fragments. The purpose of Hi-C is to understand the three-dimensional conformation of nuclear chromatin, obtain chromatin sequencing fragments that are very close in spatial position or interact with each other in the cell nucleus, so as to better study the interaction within or between chromatin, and the regulation of gene regulatory elements on a genome-wide scale. If you have a hic format file, you need to use the hicConvertFormat of the HiCexplorer software to convert the .hic/ hicpro matrix file into the ginteractions format before using this function, and then convert it into a three-column format file of txt or csv. You can also use the Straw tool.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see below
- Set the X-axis and Y-axis on and off, font, font size, color, rotation angle and step length
- Scale-min and Scale-max can customize the value range of the Color bar, or you can select None and let the system automatically define it.
- show data can display the data value of each square on the heat map, and can also adjust the display font size of the data value.
- Scale label is divided into out, into, and in, which adjusts the orientation of the label teeth of the X and Y coordinate axes.
- Cell square converts each small square generated by the heat map into a square.
- Color bar sets its direction, label font size, label name, size, and color.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Line widths is the dividing line of the heat map. If necessary, you can adjust the color and select a value greater than 0 for the width.
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
TAD
The genome interaction map is essentially a symmetrical matrix, with equal information on both sides of the diagonal. The interaction strength changes from weak to strong, and the color of the cell changes from white to red. There are repeated small triangular areas on the bottom edge, and the interior is almost entirely red, indicating that the interaction frequency between the chromatin fragments in these areas is high. Such areas are called self-interaction areas, while the interaction frequency between adjacent triangular areas is low. The red triangular areas correspond to the interaction information of the internal areas of TAD, and the black areas correspond to the interaction information between TADs. Presented on the triangular interaction map, there are many small red triangles on the bottom edge, and the interaction areas corresponding to the triangles are all white. Scientists define this domain with high internal interaction frequency and low inter-group interaction frequency as topologically associated domain, referred to as TAD. If you have a hic format file, you need to use the hicConvertFormat of the HiCexplorer software to convert the .hic/ hicpro matrix file into ginteractions format before using this function, and then convert it into a three-column format file of txt or csv.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see below
- Set the X-axis and Y-axis on and off, font, font size, color, rotation angle and step length
- Scale-min and Scale-max can customize the value range of the Color bar, or you can select None and let the system automatically define it.
- show data can display the data value of each square on the heat map, and can also adjust the display font size of the data value.
- Scale label is divided into out, into, and in, which adjusts the orientation of the label teeth of the X and Y coordinate axes.
- Cell square converts each small square generated by the heat map into a square.
- Color bar sets its direction, label font size, label name, size, and color.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Line widths is the dividing line of the heat map. If necessary, you can adjust the color and select a value greater than 0 for the width.
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
Chr density plot
The Chromosome Density Tool is a drawing tool used to represent the interactions between chromosomes of different species and between chromosomes of the same species. This helps to study the functional associations between chromosomes.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see below
Detailed instructions:
- Set the X-axis and Y-axis on and off, font, font size, color, rotation angle and step length
- Scale-min and Scale-max can customize the value range of the Color bar, or you can select None and let the system automatically define it.
- Scale label is divided into out, into, and in, which adjusts the orientation of the label teeth of the X and Y coordinate axes.
- Color bar sets its direction, label font size, label name, size, and color.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Title and Y-axis label set input text
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 300-1000dpi 4-level images for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
Chr. circle plot
Chr. circle plot can be used for many types of data, including heat maps, bar graphs, line graphs, and scatter plots. It is suitable for gene expression data, proteome data, genome data, chromatin interaction data, DNA methylation data, ChIP-seq data, transcriptome data, epigenomic data, mutation and structural variation data.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see below
Detailed instructions:
- In Select file, input the drawing file, Select file on length, input the chromosome length file, Select file on circle1-4, input the data for each circle, Select file on Links, input the chromosome colinearity data.
- Set Tick on/off and font size
- Color map 1-4 contains 166 types, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Color bar label 1-4 corresponds to the color bar labels of the four circles
- Color bar sets the label font size and scale font size.
- The Scatter type 'Shape' sets 15 point shapes, and the 'Color' parameter sets the color
- The Bar type sets the color using the 'Color' parameter and the 'Line style' parameter sets the 4 line types.
- Line type sets the color and line width parameters
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 202 3. 12.25
Manhataan
In genome association studies (GWAS), Manhattan plots are used to display the association strength of each locus in the genome. By plotting statistical significance (such as p-values) against genomic location, researchers can visually identify genetic variants associated with specific traits or diseases. This facilitates the integrated analysis of genomic data and phenotypic data in multi-omics and reveals the genetic basis of complex traits.
Detailed usage:
1. First click the Upload button in the Upload data area to enter the file to be processed or enter relevant data in the input box.
2. Enter the parameters. If you need to adjust the parameters, please see below.
- Set the X-axis and Y-axis on and off, font size, color, rotation angle and step length
- Set the label name and font size of the X-axis, Y-axis and label
- "Image resolution" parameter sets the image resolution
- " Sign marker color " sets the color of the special mark
- " Sign marker P-value " sets the P-value of the special marker
- "Dot shape" sets 23 dot shapes
- The "Transparency" parameter can adjust the transparency of the point color
- Set the legend title and title font size, legend font size, and legend position
- "Color map" contains 166 types, mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Sets the line type and width
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 4. 16
Updated: 2024. 4. 16
QQ plot
In statistical analysis and genome-wide association studies, QQ plots are used to compare the observed p-value distribution with the expected uniform distribution. By detecting deviations, researchers can identify potential systematic biases or true association signals. This helps assess data quality and verify the reliability of analysis results in multi-omics.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Set the font size, color, rotation angle and step size of the X and Y axes
- Set the label name and font size of the X-axis, Y-axis and label
- "Image resolution" parameter sets the image resolution
- " Dot shape " sets the shape of 23 types of dots
- " Dot color " sets the color of the dot
- " Datum line color " sets the color of the baseline
- " Transparency " parameter can adjust the transparency of the point color
- Set the legend title and title font size, legend font size, and legend position
- "Color map" contains 166 types, mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 4. 16
Updated: 2024. 4. 16
SSR prediction
Simple sequence repeat (SSR) prediction tools are used to identify microsatellite sequences in the genome. SSR is a highly polymorphic molecular marker that is widely used in genetic diversity analysis, genome map construction, and gene function research. In multi-omics studies, SSR prediction tools help researchers understand the structural variation and evolutionary mechanisms of the genome.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Online time: 2024.6.13
Update time: 2024.6.13
BLAST
The Basic Local Alignment Search Tool (BLAST) is used to quickly find similar sequences in a database and identify the functions and evolutionary relationships of genes or proteins through sequence alignment. In multi-omics studies, BLAST is a key tool for annotating new sequences, identifying conserved gene families, and comparing gene homology between different species.
Detailed instructions:
- First, click the input box area to enter the relevant data that needs to be processed
- Select the species file you want to compare, and you can also enter related advanced settings
2. After completing all configurations, click the " BLAST " button to start running.
3. The output results are displayed in the Result area.
Online time: 2024.6.13
Update time: 2024.6.13
2.4 Evolution
Density peak plot
Ridge plot is also called peak plot or mountain plot. It mainly displays the same X-axis data, which can be time series, genetic data, etc., corresponding to different Y-axis data, to clearly show the relationship between different data and variables.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see below
Detailed instructions:
- Set the X-axis and Y-axis on and off, font, font size, color, and rotation angle
- The X-axis label number parameter is used to set the number of x-axis labels.
- Color bar sets its direction, label font size, label name, size, color, position, and font size.
- There are 166 types of color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Vertical spacing sets the distance between sub-images
- The Y-axis label sets the rotation direction and font size
- X-axis label sets the input text and font size
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
Evolutionary tree
Evolutionary tree visualization tools are essential in multi-omics studies. They illustrate evolutionary relationships among species or genes, integrating data from genomics, proteomics, transcriptomics, and metabolomics. This helps researchers understand functional genomics, trace trait evolution, and infer evolutionary pressures. These tools also allow for the comparison of phylogenetic trees from different omics layers, enhancing our understanding of the interplay between genetic, epigenetic, and environmental factors.
Detailed instructions:
1. First, select the input file format, and then click the "Upload" button in the "Upload data" area to input the files that need to be processed.
2. Input parameters. If you need to adjust the parameters, please see below.
- The font can be adjusted in type, size and color.
- Layout the overall shape of the evolutionary tree
- Whether the Branch length is enabled
- Tree style sets the shape, width and color of the clustering tree
- The Color bar sets the on and off as well as the label name.
- Set the shape, size, transparency and color of the parameter points
- Width and height parameters have been set
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 2024.7.29
Update time: 2024.7.29
2.5 Gene editing
GuideRNA
Design guide RNA for CRISPR-Cas system for gene editing to help achieve precise gene knockout or repair.
Detailed instructions:
1. First, click " Select Genome " and " Choose genome annotation file (.gtf) " in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- "Custom PAM (Max 6bp)" parameter is an optional parameter for setting a custom PAM for the sgRNA
- "Call Off-Targets?" parameter is TRUE, the function will search for off-targets in the genome specified by the genomename parameter. If it is FALSE, off-target calls will be skipped.
- "Annotate Off-Targets?" parameter is TRUE, the function will provide annotations for off-targets called using the genomic annotation file specified by the gtfname parameter. If FALSE, off-target annotations will be skipped.
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time : 202 3.3.7
Update time: 2023.3.7
Primer design
Used to design PCR primers to assist in DNA amplification, cloning, and sequencing in experiments.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- The maximum number of primers (pairs) returned by the "Number To Return" parameter
- "Primer Size" parameter inputs the primer length
- "Primer Tm" parameter to enter the optimal TM value
- "Primer GC" parameter inputs the percentage of GC content
- "Product Size" parameter inputs the length range of the primer product
- "Thermodynamic oligo alignment" parameter is set to 1, the program will use a thermodynamic model to calculate the probability of oligos forming hairpin structures and dimers.
- "Max Poly-X" parameter enters the maximum number of mononucleotide repeats allowed
- "Internal Max Poly-X" parameter inputs the number of mononucleotide repeats allowed
- "Salt monovalent" parameter input PCR reaction system monovalent salt concentration
- The "DNA conc" parameter enters the concentration of the annealing oligo after PCR.
- "Max self any" parameter inputs the probability of the primer self-binding
- The "Pair Max compl any" parameter inputs the probability of the left primer binding to the right primer
- "Pair Max compl end" parameter Input the 3' end complementary pair of left primer and right primer
- "GC clamp" parameter Enter the 3' end sequence of the left primer and the right primer with a specified number of consecutive Gs or Cs bases.
- "Max NS accepted" parameter inputs the number of N bases allowed
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 2023.12.25
Update time: 2023.12.25
2.6 Format conversion
Seq format convert
Convert different sequence file formats, such as from FASTA to FASTQ, to facilitate data processing and analysis.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Online time: 2023.12.25
Update time: 2023.12.25
Vcf2Phylip
Convert VCF format mutation data into Phylip format for phylogenetic tree construction and evolutionary analysis.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.6. 24
Update time: 2024.6. 24
Tree format convert
Convert different phylogenetic tree formats, such as from Newick to Nexus, to facilitate data compatibility between different software.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.6. 24
Update time: 2024.6. 24
Bam2Fasta
Convert BAM format files to FASTA format to facilitate further analysis and processing of sequence data .
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.6. 24
Update time: 2024.6. 24
Bam2Fastq
Convert BAM format files to FAST Q format to facilitate further analysis and processing of sequence data .
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.6. 24
Update time: 2024.6. 24
Bam2Sam
Convert BAM format files to SAM format to facilitate further analysis and processing of sequence data .
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.6. 24
Update time: 2024.6. 24
Json2Xml
JSON and XML formats to facilitate data sharing and processing.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.6. 24
Update time: 2024.6. 24
Json2Fasta, Fastq
JSON converts between FASTA, FASTQ and other formats to facilitate data sharing and processing.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.6. 24
Update time: 2024.6. 24
Xml2Fasta, Fastq
XML converts between FASTA, FASTQ and other formats to facilitate data sharing and processing.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.6. 24
Update time: 2024.6. 24
3.1 Transcription
Sequence conversion
Translating nucleic acid sequences into protein sequences helps study the proteins encoded by genes and their functions.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Online time: 2023.12.25
Update time: 2023.12.25
3.2 Heatmaps
Heatmap
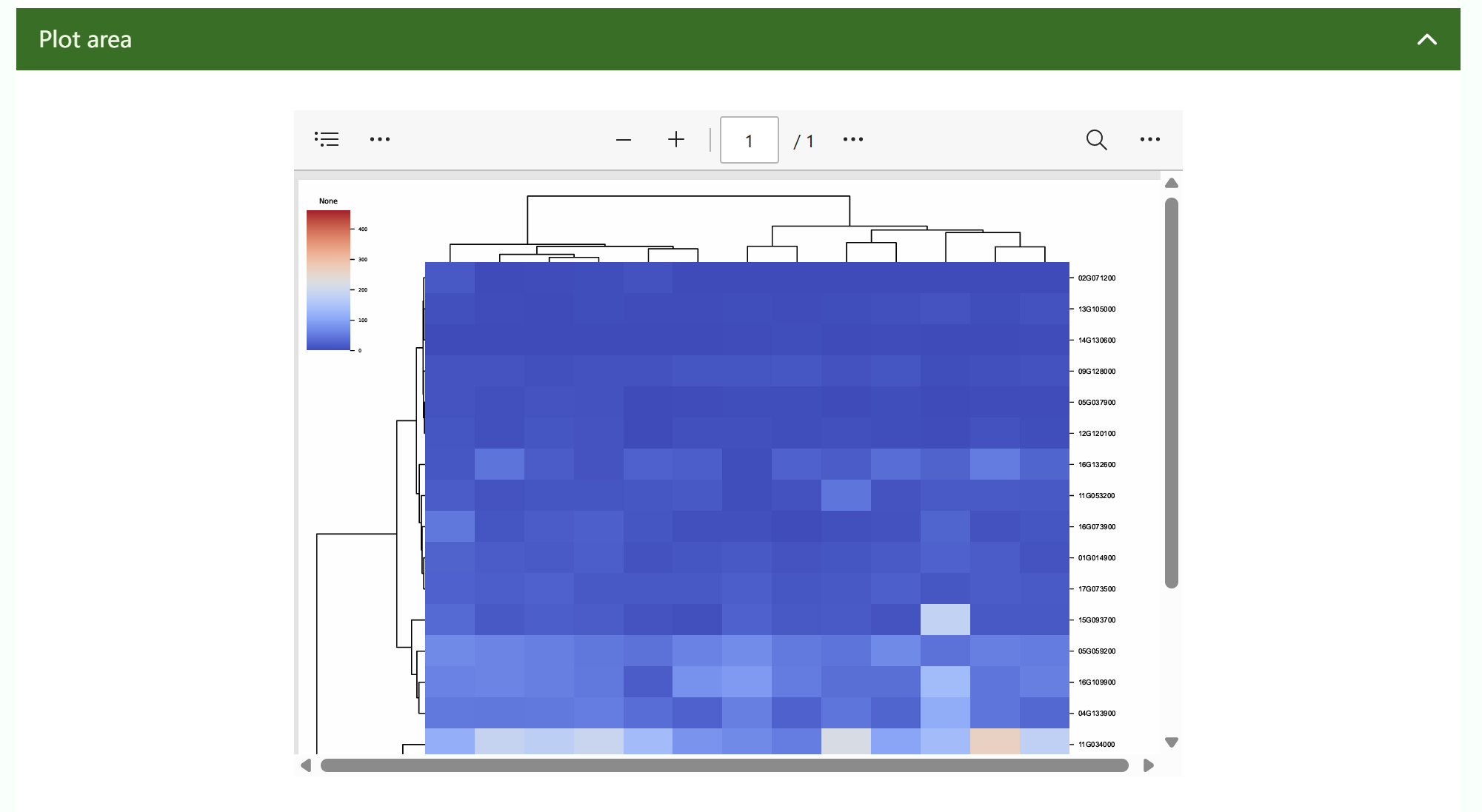
Heatmap can draw ordinary heatmaps or heatmaps with tree-like clustering, which can be used by users who want to cluster data. This type of map can be simply understood as using a distance algorithm on the original basic heatmap to cluster data values with similar distances into one category. 7 clustering methods and 22 distance metrics are provided.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- X-axis and Y-axis set the opening and closing, font, font size, color, rotation angle and step size of the X-axis and Y-axis.
- Row cluster and Col cluster control the opening and closing of the row and column clustering dendrograms.
- The Normalization parameter normalizes the heat map. You can choose between Standard and Z-score, and select row or column processing.
- Scale-min and Scale-max can customize the value range of the Color bar, or you can select None and let the system automatically define it.
- show data can display the data value of each square on the heat map, and can also adjust the display font size of the data value.
- Metric can choose 7 clustering methods
- Method can choose 22 distance metrics
- Cell square converts each small square generated by the heat map into a square.
- Color bar sets its direction, label font size, label name, size, and color.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Line widths is the dividing line of the heat map. If necessary, you can adjust the color and select a value greater than 0 for the width.
- Column comment can open column comments. Enter the name of the column to be commented on in Comment column name, which is also the name of the comment column in the figure.
- Classify color1, Classify color2, Classify color3, what color should I choose
- Tree style sets the shape, width, and color of the clustering tree.
- Robust is turned on to use quantiles to calculate the color map range
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 300-1000dpi 4-level images for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
Circle heatmap
Circle heatmap, as the name implies, is a form of heatmap. The advantage of circle heatmap is that it can show multiple aspects in one picture. It is suitable for multi-group or multi-omics research and can reveal the changing patterns and connections of different omics.
Detailed instructions:
- First, click the file button on the right and select the Csv format or Excel format file that needs to be visualized
- The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- The Cluster by parameter is a clickable button that will jump to the corresponding clustering tool.
- Set the on/off, font, font size, color, and rotation angle of the X-axis and Y-axis.
- show data can display the data value of each square on the heat map, and can also adjust the display font size of the data value.
- The Color bar sets its direction, label font size, label name, height and width, and color.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Border widths is the dividing line of the heat map. If necessary, you can adjust the color and select a value greater than 0 for the width.
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
Circle heatmap (cluster)
Circle heatmap (cluster) is a heatmap mode that combines circle heatmap with tree clustering.
Detailed instructions:
- First, click the file button on the right and select the Csv format or Excel format file that needs to be visualized
- The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- Metric can choose 7 clustering methods
- Method can choose 22 distance metrics
- Set the on/off, font, font size, color, and rotation angle of the X-axis and Y-axis.
- show data can display the data value of each square on the heat map, and can also adjust the display font size of the data value.
- The Color bar sets its direction, label font size, label name, height and width, and color.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Border widths is the dividing line of the heat map. If necessary, you can adjust the color and select a value greater than 0 for the width.
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
Correlation matrix
Correlation matrix is used to find pairwise correlations of all columns in a data frame and visualize which variable is correlated with another variable.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- The Matrix type parameter can select whether the generated image is rectangular or triangular.
- Set the on/off, font, font size, color, rotation angle and step size of the X-axis and Y-axis.
- Scale-min and Scale-max can customize the value range of the Color bar, or you can select None and let the system automatically define it.
- show data can display the data value of each square on the heat map, and can also adjust the display font size of the data value.
- Scale label is divided into out, into, and in, which adjusts the orientation of the label teeth of the X and Y coordinate axes.
- Cell square converts each small square generated by the heat map into a square.
- Color bar sets its direction, label font size, label name, size, and color.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Line widths is the dividing line of the heat map. If necessary, you can adjust the color and select a value greater than 0 for the width.
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
3D heatmap
3D heatmap can display data in three dimensions at the same time. Its appearance makes complex data analysis results clearer and can easily obtain data analysis results directly from the graph.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- X-axis, Y-axis, and Z-axis set the on/off, font, font size, color, and rotation angle of the X-axis, Y-axis, and Z-axis.
- Color bar sets its direction, label font size, label name, size, and color.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- The Cluster by parameter is a clickable button. You can select row clustering and column clustering, and it will jump to the corresponding clustering tool.
- Number of color bar scales Set the number of scales.
- Bar label and Bar label size can set the label and font size of the Color bar
- Title, X-label, Y-label, Z-label can set labels
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 300-1000dpi 4-level images for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
3D cluster by Row
3D cluster by Row performs column clustering based on the 3D heat map. It provides 7 clustering methods and 22 distance metrics.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- X-axis, Y-axis, Z-axis set the opening and closing, font, font size, color, and rotation angle of the X-axis, Y-axis, and Z-axis.
- Metric can choose 7 clustering methods
- Method can choose 22 distance metrics
- Color bar sets its direction, label font size, label name, size, and color.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Number of color bar scales Set the number of scales
- Bar label and Bar label size can set the label and font size of the Color bar
- Title, X-label, Y-label, Z-label can set labels
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
3D cluster by Col
3D cluster by Col performs row clustering based on the 3D heat map. It provides 7 clustering methods and 22 distance metrics.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see 3D cluster by Col parameters
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
3D scatter plot
3D scatter plot is very similar to 3D heat map. All data are represented by cube scattered points in three-dimensional coordinates, and colors are represented by corresponding two-dimensional data. The most distinctive feature is that the three-dimensional heat map is very similar to the two-dimensional heat map when viewed from a bird's-eye view, ensuring the accuracy of the heat map. The third type of data can be represented by color differences or by the height in three-dimensional space.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see 3D plot parameters
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
3D bar plot
3D bar plot is similar to 3D scatter plot . All data are represented by cylinders in three-dimensional coordinates. The colors are represented by the corresponding data in two dimensions. The data can be represented by color differences or by the height of the cylinders.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see 3D plot parameters
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 300-1000dpi 4-level images for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
2.3 . 7.1 Isoheight plot
Isoheight plot is a heat map mode that we combine with contour map and tree clustering. Contour heat map uses two-dimensional form to reflect three-dimensional data. This type of heat map is more suitable for a large amount of biological data. Through color differences, we can see the differences between data at a macro level.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- X-axis and Y-axis set the opening and closing of X-axis and Y-axis, font, font size, color, rotation angle and step size
- The Cluster by parameter is a clickable button that will jump to the corresponding clustering tool.
- Row cluster and Col cluster control the opening and closing of the clustering dendrogram of rows and columns
- Scale-min and Scale-max can customize the total value range of the Color bar, and Scale interval sets the value range of each color block on the Color bar. You can change the fineness of the color to adjust the drawing effect.
- Show data can display the data value of each square on the heat map, and can also adjust the display font size of the data value.
- Color bar sets its direction, label font size, label name, size, and color.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Number of color bar scales Set the number of scales
- Bar label and Bar label size can set the label and font size of the Color bar
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
2.3 . 7.2 Isoheight plot (cluster)
Isoheight plot (cluster) is a heat map mode that combines contour map with tree clustering. Contour heat map uses two-dimensional form to reflect three-dimensional data. This type of heat map is more suitable for a large amount of biological data. It surrounds high expression values by clustering. Through color differences, we can see the difference between data at a macro level. The clustering effect is reflected by a variety of tree colors, which is also one of the highlights of contour heat map.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- X-axis and Y-axis set the opening and closing of X-axis and Y-axis, font, font size, color, rotation angle and step size
- Row cluster and Col cluster control the opening and closing of the clustering dendrogram of rows and columns
- Scale-min and Scale-max can customize the total value range of the Color bar, and Scale interval sets the value range of each color block on the Color bar. You can change the fineness of the color to adjust the drawing effect.
- Show data can display the data value of each square on the heat map, and can also adjust the display font size of the data value.
- Metric can choose 7 clustering methods
- Method can choose 22 distance metrics
- Color bar sets its direction, label font size, label name, size, and color.
- There are 166 types of color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Number of color bar scales Set the number of scales
- Bar label and Bar label size can set the label and font size of the Color bar
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 300-1000dpi 4-level images for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
Dot heatmap
hbiCloud adds the Dot plot function, which can draw heat maps using 23 different shapes such as point, diamond, circle, star, etc., to increase the richness of heat map graphics. In addition to the color representing the data, the size of the dot and the depth of the color are consistent, and they represent the size of the value at the same time. And the points can be zoomed in and out synchronously.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see below
Detailed instructions:
- Set the X-axis and Y-axis on and off, font, font size, color, rotation angle and step length
- Color bar sets its direction, label font size, label name, size, color, position, and font size.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- " Dot shape " sets 23 dot shapes
- " Dot size magnification " can adjust the size of the dot
- " Transparency " parameter can adjust the transparency of the point color
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12 . 25
Update time: 2023.12 . 25
Anatomogram plot
Effectively displaying tissue information in multicellular organisms is a time-consuming and laborious process. In anatomical diagrams, using different colors to represent the expression of tissues makes it easy to find differences between tissues or tissues, and immediately provides the biological context of these observations, allowing for faster grasp of visualization results.
Detailed instructions:
1. First click the file button on the right and select the Excel or Csv format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- TypeSelect the required anatomical base map
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Color bar sets its direction selection
- Title, label, Color bar can set label name
3. After completing all configurations, click the "Run" button to display or update the Anatomogram plot
, and it appears in the canvas on the left.
- Users can export the heat map by right-clicking and selecting the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000 dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
3.3 Expression conversion
RPKM2TPM, TMM
These tools are used to normalize RNA sequencing data. The conversion of RPKM (Reads Per Kilobase Million) to TPM (Transcripts Per Million) and TMM (Trimmed Mean of M-values) can correct for differences in sequencing depth and gene length, making expression levels between different samples more comparable.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Online time: 202 4 . 6.13
Update time: 202 4 . 6.13
3.4 WGCNA
Sample cluster
Used for sample cluster analysis to identify similarities and differences between samples.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- The maximum number of primers (pairs) returned by the "Method" parameter
- "Text size" parameter sets the size of the inner label
- The "Axis size" parameter sets the axis scale font size
- "X-label" parameter and the "Title" parameter set the label name
- "X-label size" parameter and the "Title size" parameter set the label name size
- "Tree width" parameter sets the width of the tree
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 202 4 . 5.21
Update time: 202 4 . 5.21
Diagnostic plot
Used to diagnose data quality and detect outliers.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- The "Text color" parameter sets the inner label color
- "Abline color" parameter sets the horizontal line color
- The "Network type" parameter sets the network type
- "Max" parameter powers the maximum value
- "Increment" parameter powers the value of the increase
- The "Verbose" parameter sets the integer level of verbosity
- "Text size" parameter internal label size
- Set the width and height parameters
- Set the coordinate labels and main label names of the two output images
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 202 4 . 5.21
Update time: 202 4 . 5.21
Cluster dendrogram
Generate a cluster tree to display the hierarchical clustering results of genes or samples.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- The "Network type" parameter sets the network type
- The "Power" parameter sets the soft threshold capability of the network construction
- "TOMType" parameter sets the TOM type
- The "Pam respects dendro" parameter is only used if pamStage is TRUE. If TRUE, the PAM stage will respect the logic of the dendrogram, i.e. objects can only be assigned to clusters below the branch they merged.
- The "Save TOMs" parameter sets whether the vertex overlap matrix of each module should be saved and returned
- The "Save TOM file base" parameter sets the string containing the base name of the consistent topology overlay file
- The "Deep split" parameter provides simplified control over how sensitive module detection is to module splits.
- The "Reassign threshold" parameter sets the threshold for the p-value ratio of genes between reassigned modules.
- The "Min module size" parameter sets the integer level of detail
- The "Max block size" parameter sets the maximum module detection block size as an integer
- The "Numeric labels" parameter specifies whether the returned modules should be labeled with colors or numbers.
- The "Merge cut height" parameter sets the cut height of the tree view when merging modules
- The "Verbose" parameter sets the verbosity level
- "Dendro labels" parameter sets the dendrogram labels
- The "Add guide" parameter specifies whether vertical "guide lines" should be added to the dendrogram.
- The "Guide Hang" parameter is the fraction of the dendrogram height that is left between the top of the guideline and the combined height of the dendrogram.
- The "corType" parameter sets the string of correlations to use
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 202 4 . 5.21
Update time: 202 4 . 5.21
Module-trait
Analyze the relationship between modules and traits to help identify gene modules associated with specific biological characteristics.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- "X-font rotation" parameter X-axis scale rotation angle
- The "Text size" parameter sets the internal label font size
- The "Set standard margin" parameter sets whether to set the standard margin before calling the drawing function
- The "Color labels" parameter sets whether the xlabel and ylabel are interpreted as colors
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 202 4 . 5.21
Update time: 202 4 . 5.21
4.1 Sequence & Structure
Protein analysis
Including the number of amino acid occurrences, molecular weight, aromaticity, instability index, isoelectric point, helical fraction, number of turns, number of plates and molar extinction coefficient, to help study the biological functions and interactions of proteins.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Online time: 2023.12.25
Update time: 2023.12.25
DNA2Protein
Translating DNA sequences into protein sequences helps study the proteins encoded by genes and their functions.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Online time: 2023.12.25
Update time: 2023.12.25
2nd Struct. Domain
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- " Feature " parameter selects whether a feature file needs to be input
- " Feature file " parameter input feature file
- " Mark shape " parameter selects the shape of the mark
- " Aligning " parameter is aligned, input alignment gene
- " Element label " parameter sets whether to display element labels
- Set the width and height parameters
- "Color map" parameter contains 166 types, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- and label size of the X and Y axes
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 202 4 . 6.13
Update time: 202 4 . 6.13
3D Struct
By predicting and visualizing the 3D structure of proteins, we can study their functions and interactions and understand their roles in biological processes.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- " alpha helix color " parameter sets the alpha helix color
- " β-sheet color " parameter sets the β-sheet color
- The " Cartoon color " parameter sets the structure color
- The " Background color " parameter sets the background color
- " Axis of rotation " parameter sets the axis around which the rotation occurs.
- " Angle " parameter rotates the given axis by the given angle.
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 202 4 . 5.8
Update time: 202 4 . 5.8
Signal peptide prediction
Predict signal peptide sequences and identify whether a protein has a signal peptide that directs its secretion or localization.
Launch time: 2024.6. 24
Update time: 2024.6. 24
4.2 Difference & Enrichment
Enrichment bar plot
Enrichment bar plot is a multivariate histogram . The correlation between data can be analyzed by the position, height and color of the bar . It is suitable for data with small amount of data and clear position relationship. GO and KEGG enrichment data are common usage scenarios of enrichment bar plot r. The color of the bar represents the p-value (or q-value, etc.), and the size represents the number of genes.
Detailed instructions:
1. First click the file button on the right and select the Excel format or Csv format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- Set the X-axis and Y-axis on and off, font, font size, color, rotation angle and step length
- Color bar sets its direction, label font size, label name, size, color, position, and font size.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- The Transparency parameter can adjust the transparency of the point color
3. After completing all configurations, click the "Run" button to display or update the bubble plot , and the bubble plot will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
Bubble plot
Bubble plot is a multivariate chart and a variation of scatter plot. Bubble plot is a combination of scatter plot and percentage area chart. The correlation between data can be analyzed by the position, area and color of the bubble . It is suitable for data with small amount and clear position relationship. GO and KEGG enrichment data are common use scenarios of bubble plot. The color of the bubble represents the p value (or q value, etc.), and the size represents the number of genes.
Detailed instructions:
1. First click the file button on the right and select the Excel format or Csv format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- Set the X-axis and Y-axis on and off, font, font size, color, rotation angle and step length
- Color bar sets its direction, label font size, label name, size, color, position, and font size.
- Bar lable2 is used as the label of the size bar, and the Bar lable2 size parameter is added. The 'Distance between entries in the legend' parameter can set the internal label distance of the size bar
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Dot shape sets 15 dot shapes
- Dot size magnification can adjust the size of the dot
- The Transparency parameter can adjust the transparency of the point color
3. After completing all configurations, click the "Run" button to display or update the bubble plot , and the bubble plot will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
upsetR
A tool for set visualization that can show the intersection and union relationships between multiple data sets and provide more detailed interaction information than the traditional Venn diagram.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- " Point size " parameter sets the point size
- " Line width " parameter sets the width of the line
- " Type " parameter sets the type of point
- " Main bar color " parameter sets the color of the bar chart
- " Matrix color " parameter setting allows to define the color of the intersection points
- The "Data rotation" parameter sets the rotation angle of the internal labels
- The "Keep order" parameter can be set to display in the order of the provided data.
- Sets the axis label name parameter
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Online time: 2024.3.7
Update time: 2024.3.7
Treemap
Treemap, also known as rectangular tree structure drawing method, also known as rectangular tree structure diagram drawing method, tree structure rectangular diagram drawing method, refers to a method of using nested rectangles to display tree structure data. This presentation method can present different categories in different color blocks, and the size of each category can be compared through the size of the block. The larger the block range, the larger and more the value of the category.
Detailed instructions:
1. First click the file button on the right and select the Excel format or Csv format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- Show data can display the data value of each square on the heat map, and can also adjust the display font size of the data value.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Border widths is the dividing line of each small grid of Treemap . If necessary, you can adjust the color and select a value greater than 0 for the width.
- Transparency sets the transparency of the color block, ranging from 0 to 1.
- Title can set labels and font size
3. After completing all configurations, click the "Run" button to display or update the Treemap, and the Treemap will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
Wordcloud
Wordcloud, also known as word cloud, is a visual representation of text data. It is a colorful cloud-like graphic composed of words and is used to display large amounts of text data. The importance of each word is displayed in font size or color. Wordcloud plot is mainly used to analyze the expression value of a term, and is suitable for visualizing terms with high expression values. Words with higher values in the word cloud will be presented in a larger form, and words with lower values will be presented in a smaller form. The essence of a word cloud is a point map, which is the result of drawing text with a specific style at the corresponding coordinate points.
Detailed instructions:
1. First click the file button on the right and select the Excel format or Csv format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- Width and Height set the width and height of the output image
- Background color parameter selects the background color
- Word distance sets the distance between words
- Maximum word count and Maximum font size can customize the total value range of the font size
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- 'The color is the same as the background image color' asks whether the font color of the output image is consistent with the background image
3. After completing all configurations, click the "Run" button to display or update the Wordcloud map, and the Wordcloud map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12.25
Update time: 2023.12.25
Venn
Used to draw Venn diagrams to show the overlap and unique parts between multiple data sets, helping to understand the common and unique elements between different groups.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Color map contains 166 types, mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is the horizontal flip of the original color bar. The "Font size" parameter sets the font size
- The " Image resolution " parameter sets the image resolution
- " Number " parameter sets whether to display the value
- " logic " parameter sets whether to display dependencies
- " percent " parameter sets whether to display percentages
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Online time: 2024.3.7
Update time: 2024.3.7
KEGG PathView
Used for visualization of KEGG pathway diagrams, combined with gene expression or metabolic data, to show the role and changes of genes in specific biological pathways.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- " Species " parameter enters the abbreviation of the species in the KEGG library. For details, please refer to the link https://www.genome.jp/kegg/tables/br08606.htm
- " Gene id type " parameter sets the character ID type used for gene.data, case-insensitive
- " Output suffix " parameter sets the suffix to be added to the output image file as a path name suffix
- The " Pathway id " parameter sets the KEGG pathway ID
- " Kegg native " parameter is TRUE, which outputs a png file of the complete pathway. Otherwise, it outputs only a pdf file of the input gene list.
- " Expand node " parameter sets whether a multi-gene node should be expanded to a single-gene node
- " Split group " parameter sets whether to split the node group into separate nodes .
- " Map symbol " parameter sets whether to map gene IDs to gene node labels using the symbol or the graphic name in the KGML file.
- " New signature " parameter sets whether the path view signature is added to the path view .
- " Min nnodes " parameter is the minimum number of nodes of type "gene", "enzyme", "compound" or "homolog" required to be considered a pathway
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Online time: 2024.6.13
Update time: 2024.6.13
GSEA
The GSEA (Gene Set Enrichment Analysis) tool is important in multi-omics for identifying enriched biological pathways or gene sets in large datasets. It helps uncover functional patterns and key biological processes associated with different conditions, supporting a deeper understanding of molecular mechanisms in integrated omics studies.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- The "gene_sets" parameter can set the built-in gene set library
- The "threads" parameter sets the number of threads
- The parameters min_size and max_size are set to maximum and minimum
- The Permutation_num parameter sets the number of permutations
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 2024.10.9
Update time: 2024.10.9
Format conversion
The Metabolic Data Format Conversion tool plays a crucial role in multi-omics research by enabling the seamless conversion between various data formats. This tool supports a wide range of formats including mzML, mzXML, IdXML, mzIdent, pepXML, protXML, featureXML, consensusXML, and traMLF. These formats are commonly used in metabolomics and proteomics for storing mass spectrometry data, identification results, peptide and protein information, feature extraction, and consensus data. By facilitating the interoperability between these formats, the tool allows researchers to integrate and analyze complex datasets more efficiently, thereby enhancing the robustness and comprehensiveness of multi-omics studies.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- The "Input file fomat" parameter inputs the format of the target file
- The "Start" parameter is used to input the sequence position where extraction begins
- The "Output file fomat" parameter outputs the format of the target file
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 2024.8.1
Update time: 2024.8.1
Scatter and PCA plot
hbiCloud adds the Scatter plot function, which is a distribution diagram of data points on a rectangular coordinate plane. You can use 23 different shapes such as point, diamond, circle, star, etc. to draw the diagram. In addition to the color representing the data, the size of the point and the depth of the color are consistent, and they represent the value at the same time. You can also achieve synchronous zooming in and out of the points.
PCA (Principal Component Analysis) is the most widely used data dimensionality reduction algorithm. The main idea of PCA is to map n-dimensional features to k-dimensional features. These k-dimensional features are new orthogonal features also called principal components. They are k-dimensional features reconstructed based on the original n-dimensional features.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see below
Detailed instructions:
- The PCA parameter is used to select whether to perform PCA calculation on the data, and the Number of feature dimensions parameter is used to select the number of feature dimensions to reduce the data.
- Set the X-axis and Y-axis on and off, font size, color, rotation angle and step length
- Color bar sets its direction, label font size, label name, size, color, position, and font size.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Dot shape sets 23 dot shapes
- Dot size magnification can adjust the size of the dot
- The Transparency parameter can adjust the transparency of the point color
3. After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export heat maps by right-clicking the "Save" export option. hbiCloud provides 4-level images of 300-1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 202 3. 12.25
Update time: 202 3. 12.25
Volcano plot
Plot a volcano plot to help identify significantly differentially expressed genes by displaying gene expression changes (log fold change) and statistical significance (-log10 p-value).
Detailed usage:
1. First click the Upload button in the Upload data area to enter the file to be processed or enter relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- " Dot size" parameter sets the dot size
- The "Number up" and "Number down" parameters set the number of up-regulated genes and the number of down-regulated genes
- "Gene size" parameter sets the size of gene names
- The "Fdr" parameter sets the false positive rate
- "Logfc" parameter setting distinguishes up- and down-regulated genes
- The "X threshold" parameter sets the X-axis threshold
- Set the label name and font size of the X-axis, Y-axis and label
- Label names and colors of the up-regulated group, down-regulated group, and unchanged group
- Set the width and height parameters
- The "Alpha" parameter sets the transparency
- The "Line type" parameter sets the line type
- The "Gene color" parameter sets the color of the gene name
- "Fontface" parameter gene name style
- The "Box unit x" parameter and the "Box unit units" parameter set the amount of padding around the bounding box
- " Point unit x" parameter and the " Point unit units" parameter set the amount of padding around the marker point
- " Segment colour " parameter sets the colour of the line segments connecting the points and labels.
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Online time: 2024.3.7
Update time: 2024.3.7
Network plot
The Network plot function is a graphical model , which is composed of two factors : lines and nodes. Different nodes that are related are connected by one or more lines , and the node size is plotted according to the number of correlations. hbiCloud adds six different node layout methods, including random, graphic and algorithmic layouts, to give users a better custom node experience. It is often used in differential co-expression network analysis to show the correlation between different genes.
Detailed instructions:
1. First click the file button on the right and select the Excel format or Csv format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- Set the font size and color of the node.
- Color bar sets its direction, label font size, label name, size, color, position, and font size.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Nodes size magnification can adjust the size of the points.
- The Transparency parameter can adjust the transparency of the point color.
- The Nodes arrangement parameter adds six different node layouts , including random, graphical, and algorithmic layouts .
3. After completing all configurations, click the "Run" button to display or update the Network plot , and the Network plot will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12 . 25
Update time: 2023.12 . 25
Violoin plot
Combining box plots and density plots to display data distribution and its probability density helps study the differences in gene expression or metabolite levels under different conditions.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Set the font size, color, rotation angle and step size of the X and Y axes
- Set the label name and font size of the X-axis, Y-axis and label
- "Image resolution" parameter sets the image resolution
- If the "split" parameter is set to True, a half violin will be drawn for each color. This makes it easier to directly compare the distributions.
- The "inner" parameter controls the representation of the data points inside the violin plot
- The "hue" parameter sets the main grouping variable
- The "scale" parameter is used to scale the width of each violin plot.
- " bw " parameter sets the built-in variable value or floating point scaling factor used to compute the bandwidth of the kernel density.
- ' cut ' parameter sets the distance, in units of bandwidth size, to control how densely the violin plot shell extends beyond the interior extreme data points.
- Set the legend title and title font size, legend font size, and legend position
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Online time: 2024.3.7
Update time: 2024.3.7
Corrplot
It is used to display the correlation matrix between variables, indicating the correlation coefficient by color and size, and helping to identify the relationship between genes, proteins or metabolites.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- "Color map" parameter adjusts the image color
- The "Font size" parameter sets the font size
- The "Order" parameter sets the ordering method of the correlation matrix
- The "Clust method" parameter is the clust method used during cleaning.
- The "Diagonal line" parameter sets whether to display the correlation coefficient on the main diagonal line
- The "Percent" parameter sets whether to convert coefficients to percentagestyle to save space
- The "Correlation coefficient" parameter sets whether the input matrix is a correlation matrix or an uncorrelation matrix
- "Background color" parameter sets the background color
- The "Outline" parameter sets whether the outlines of circles, squares and ellipses are drawn, or the color of these glyphs.
- The "Axis label location" parameter sets the character or logical position of the text label
- The "Axis label color" parameter sets the color of the text label
- "Text label offset" parameter sets the text label
- "Text label rotation" parameter sets the text rotation angle
- Colorbar font size, width, and digital label position
- "Add Shadow" parameter sets the shadow style of the character
- The "Shadow linewidth" parameter sets the line width of the shadow
- The "Shadow color" parameter sets the color of the shadow line
- "Significant level" parameter If the p-value in p-mat is greater than the signal level, the corresponding correlation coefficient is considered insignificant.
- How to draw confidence intervals using the "Confidence interval feature" parameter
- Sets the border color and width
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Online time: 2024.3.7
Update time: 2024.3.7
Venn_network
Combining Venn diagrams and network diagrams, the intersections and unique parts of multiple data sets are displayed, while showing the relationships between elements, facilitating the integrated analysis of multi-omics data.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Color map contains 166 types, mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is the horizontal flip of the original color bar. The "Font size" parameter sets the font size
- The " Image resolution " parameter sets the image resolution
- " Number " parameter sets whether to display the value
- " logic " parameter sets whether to display dependencies
- " percent " parameter sets whether to display percentages
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 5. 8
Updated: 2024. 5. 8
6.1 Picture recognition
Seed recognition
Through image recognition technology, biological image data is automatically analyzed to help identify the width, height and circumference of plants, flowers and seeds .
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.7.12
Update time: 2024.7.12
Seedling recognition
A tool for identifying and analyzing seedlings. It uses image processing and machine learning techniques to identify the type and growth status of seedlings and output leaf area, leaf length and width, leaf angle, main stem diameter, and plant area.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.7.13
Update time: 2024.7.13
6.2 Spatial & Temporal Analysis
Calendar plot
When analyzing the characteristics of time series data, calendar plot is a more intuitive data visualization method.
Detailed instructions:
1. First click the file button on the right and select the Excel format or Csv format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- Show data can display the data value of each square on the heat map
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Border widths are the dividing lines of each month grid in Calendar plot . If necessary, you can adjust the color and select a value greater than 0 for the width.
- 'Year label color' sets the color of the month label
- Title can set tags
3. After completing all configurations, click the "Run" button to display or update the Calendar plot , and the Calendar plot will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12 . 25
Update time: 2023.12 . 25
Geographical map
Geographical plot is a map-type heat map that can display the color value on the map in a visual way according to the numerical value of each geographic location in the data.
Detailed instructions:
1. First click the file button on the right and select the Excel format or Csv format file that needs to be visualized.
2. The default parameters have been set. If you need to adjust the parameters, please see the following introduction
- Type: Enter the map type, such as "world", "china", "Guizhou", etc.
- Width and Height set the width and height of the map
- Scale-min and Scale-max can customize the total value range of the Color bar
- Color selects whether the map has color blocks
- Color bar sets its direction selection
- Title, label, Color bar can set label name
- After completing all configurations, click the "Run" button to display or update the Geographical plot . The Geographical plot will pop up as a web page, and the user can click the download button on the web page to save the image.
Online time: 2023.12 . 25
Update time: 2023.12 . 25
6.3 Data Distribution Correlation
Joint plot
Combining scatter plots and histograms to show the relationship between two variables and their univariate distribution is suitable for studying the correlation between gene expression and other omics data.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Set the X-axis and Y-axis on and off, font size, color, rotation angle and step length
- Set the label name and font size of the X-axis, Y-axis and label
- "Image resolution" parameter sets the image resolution
- The "kind" parameter sets the drawing type
- The "space" parameter controls the spacing between the joint axis and the marginal axis
- The "ratio" parameter sets the joint axis height to the marginal axis height
- The "linewidth" parameter sets the line width
- "Add category column" parameter sets the main grouping variable
- Dot shape sets 23 dot shapes
- Dot size magnification can adjust the size of the dot
- The Transparency parameter can adjust the transparency of the point color
- Set the legend title and title font size, legend font size, and legend position
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 3. 27
Updated: 2024. 3. 27
Swarm plot
By arranging data points in a non-overlapping manner, it shows the distribution of data and helps identify inter-group differences in gene expression levels or other continuous variables.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Set the X-axis and Y-axis on and off, font size, color, rotation angle and step length
- Set the label name and font size of the X-axis, Y-axis and label
- "Image resolution" parameter sets the image resolution
- " Border color " parameter and the " Border width " parameter set the border color and width
- "Add category column" parameter sets the main grouping variable
- " Dot shape " sets 23 dot shapes
- " Dot size magnification " can adjust the size of the dot
- " Transparency " parameter can adjust the transparency of the point color
- Set the legend title and title font size, legend font size, and legend position
- "Color map" contains 166 types, mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 3. 27
Updated: 2024. 3. 27
Boxen plot
The concept of box plot is expanded to show the distribution of data in more detail, which is suitable for in-depth analysis of large-scale data.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Set the X-axis and Y-axis on and off, font size, color, rotation angle and step length
- Set the label name and font size of the X-axis, Y-axis and label
- "Image resolution" parameter sets the image resolution
- The "Border color" and "Border width" parameters set the border color and width
- "Add category column" parameter sets the main grouping variable
- "Dot shape" sets 23 dot shapes
- "Dot size magnification" can adjust the size of the dot
- " Saturation " parameter is the original saturation scale used when painting the fill color.
- " Width method " parameter Envelope value box width setting method
- " Fill " parameter is true, a solid color is used. Otherwise, a line is used.
- " K_depth " parameter is the number of levels to calculate and plot for each tail.
- " Outlier prop " parameter outliers are expected to account for the proportion of data
- Confidence threshold for extreme levels of the " Trust alpha " parameter
- " Gap " parameter is scaled down by this amount on the east axis to add gaps between dodged elements.
- Set the legend title and title font size, legend font size, and legend position
- "Color map" contains 166 types, mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 3. 27
Updated: 2024. 3. 27
Resid plot
Used to display the residuals of the regression model to help assess the fit of the model and identify outliers.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Set the X-axis and Y-axis on and off, font size, color, rotation angle and step length
- Set the label name and font size of the X-axis, Y-axis and label
- "Image resolution" parameter sets the image resolution
- The "Line color" and "Line width" parameters set the line color and width
- "Add category column" parameter sets the main grouping variable
- "Dot shape" sets 23 dot shapes
- "Dot size magnification" can adjust the size of the dot
- The "Lowess smoother" parameter is the original saturation scale used when drawing the fill color.
- The "Robust" parameter uses robust linear regression when computing the residuals.
- If the "Dropna" parameter is True, observations with missing data are ignored during fitting and plotted.
- Set the legend title and title font size, legend font size, and legend position
- "Color map" contains 166 types, mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 3. 27
Updated: 2024. 3. 27
Radar Chart
Displays relative values of multidimensional data, suitable for comparing multiple omics features under different samples or conditions.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Header label name and font size
- " Category Name " parameter sets the image resolution
- " Y max " parameter sets the main grouping variable
- " Label size " sets 23 point shapes
- " Font Color " parameter is the original saturation scale used when drawing the fill color.
- " Face Color " parameter uses a robust linear regression when computing the residuals.
- " Grid Color " parameter is True, observations with missing data are ignored during fitting and the plot is
- " Spine Color " parameter is True, observations with missing data are ignored during fitting and plotted.
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 4. 16
Updated: 2024. 4. 16
7.1 Preprocessing
Standard
A data standardization tool that converts data of different scales to the same scale, making different data sets comparable. In multi-omics studies, this helps integrate and compare data from different omics levels.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.7. 4
Update time: 2024.7. 4
Z-score
A normalization method that converts data to a standard normal distribution (mean 0, standard deviation 1). In multi-omics studies, z-score can balance the impact of different omics data, making data analysis more reliable.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.7. 4
Update time: 2024.7. 4
ComBat
A tool for removing batch effects, adjusting differences between batches through linear modeling to make data more uniform and comparable. In multi-omics research, ComBat helps integrate data from different experiments or batches and reduce the interference of batch effects on analysis results.
Detailed usage:
1. First, click the Upload button in the Upload data area to enter the required file or enter the relevant data in the input box
2. Enter the parameters. If you need to adjust the parameters, please see below
4. Display the Result in the Result area
Online time: 2024.7.6
Updated time: 2024.7.6
7.2 Batch effect correction
Mice
A multiple imputation (Multiple Imputation by Chained Equations) method for handling missing values. By imputing missing values multiple times and combining the results, the Mice tool helps maintain data integrity and reduce bias caused by missing values in multi-omics studies.
Detailed usage:
1. First, click the Upload button in the Upload data area to enter the required file or enter the relevant data in the input box
2. Enter the parameters. If you need to adjust the parameters, please see below
4. Display the Result in the Result area
Online time: 2024.7.5
Updated time: 2024.7.5
KNN
K-Nearest Neighbors algorithm for data imputation. By using information from similar samples to fill in missing values, KNN can provide reliable imputation results and improve data integrity when processing multi-omics data.
Detailed usage:
1. First, click the Upload button in the Upload data area to enter the required file or enter the relevant data in the input box
2. Enter the parameters. If you need to adjust the parameters, please see below
4. Display the Result in the Result area
Online time: 2024.7.5
Updated time: 2024.7.5
7.3 Integration of Multi-Omics
iCluster
A multi-omics data integration and analysis tool that uses Bayesian models and statistical methods to comprehensively analyze multi-omics data and identify common features across omics. In multi-omics research, iCluster helps researchers reveal the complexity and interconnectedness of biological systems at multiple levels.
Detailed usage:
1. First, click the Upload button in the Upload data area to enter the required file or enter the relevant data in the input box
2. Enter the parameters. If you need to adjust the parameters, please see below
4. Display the Result in the Result area
Online time: 2024.7.7
Updated time: 2024.7.7
7.4 Denoising & Dimensionality
PCA
PCA is a statistical technique used to reduce the dimensionality of data while preserving as much variance as possible. In multi-omics studies, where data from different omics layers are integrated, PCA helps in identifying patterns and relationships between these layers. By transforming the original data into a set of uncorrelated principal components, PCA simplifies the complex data structure and allows researchers to visualize and interpret the underlying patterns more easily
Detailed usage:
1. First, click the Upload button in the Upload data area to enter the required file or enter the relevant data in the input box
2. Enter the parameters. If you need to adjust the parameters, please see below
Online time: 2024.7.7
Updated time: 2024.7.7
SVD
SVD is a matrix factorization technique used to decompose a matrix into three other matrices, which represent the original data in a lower-dimensional space. In multi-omics analysis, SVD is particularly useful for identifying latent structures and relationships between different omics datasets. It helps in uncovering hidden patterns and features by decomposing the multi-omics data matrix into singular vectors and values. This can enhance the understanding of the interactions between different omics layers and improve the integration of heterogeneous data sources.
Detailed usage:
1. First, click the Upload button in the Upload data area to enter the required file or enter the relevant data in the input box
2. Enter the parameters. If you need to adjust the parameters, please see below
4. Display the Result in the Result area
Online time: 2024.7.27
Updated time: 2024.7.27
KNN
K-Nearest Neighbors algorithm for data imputation. By using information from similar samples to fill in missing values, KNN can provide reliable imputation results and improve data integrity when processing multi-omics data.
Detailed usage:
1. First, click the Upload button in the Upload data area to enter the required file or enter the relevant data in the input box
2. Enter the parameters. If you need to adjust the parameters, please see below
4. Display the Result in the Result area
Online time: 2024.7.27
Updated time: 2024.7.27
UMAP
The UMAP tool is essential in multi-omics for reducing high-dimensional data into a lower-dimensional space, enabling visualization and clustering of complex datasets. It helps researchers identify patterns, groupings, and relationships across different omics layers, facilitating the discovery of biological insights in integrated studies.
Detailed usage:
1. First, click the file button on the right and select the Csv or Excel file that you want to visualize
2. The default parameters have been set. If you need to adjust the parameters, please see the following
Detailed usage:
4. Users can export the heat map by right-clicking the "Save" export option, and hbiCloud provides the pictures of the 4 stalls of 300-1000dpi for users to use. There are four image formats: png,.jpg, pdf,.SVG to meet different needs.
Online time: 2024.8.16
Updated: 2024.8.16
T-SNE
The T-SNE tool is valuable in multi-omics for reducing high-dimensional data to visualize complex relationships and patterns across different omics layers. It helps researchers identify clusters and outliers, facilitating the exploration of molecular similarities and differences in integrated datasets. Detailed usage:
1. First, click the Upload button in the Upload data area to enter the required file or enter the relevant data in the input box
2. Enter the parameters. If you need to adjust the parameters, please see below
4. Display the Result in the Result area
Online time: 2024.9.24
Updated time: 2024.9.24
Kmeans
Kmeans
The Kmeans tool is widely used in multi-omics to cluster data into distinct groups based on similarity. It helps identify patterns, subtypes, or molecular signatures across different omics layers, aiding in the classification and interpretation of complex biological data.
Detailed usage:
1. First, click the Upload button in the Upload data area to enter the required file or enter the relevant data in the input box
2. Enter the parameters. If you need to adjust the parameters, please see below
4. Display the Result in the Result area
Online time: 2024.9.30
Updated time: 2024.9.30
Evolutionary tree
The T-test tool is crucial in multi-omics for comparing the means of different groups, helping to identify statistically significant differences in molecular features across conditions or treatments. It supports the discovery of key biomarkers and the validation of hypotheses in integrated omics studies.
Detailed instructions:
1. First, select the input file format, and then click the "Upload" button in the "Upload data" area to input the files that need to be processed.
2. Input parameters. If you need to adjust the parameters, please see below.
- Set the font size, color and rotation Angle of the X-axis and Y-axis
- Set the label names and font sizes for the X-axis, Y-axis, and labels
- The "Image resolution" parameter sets the resolution of the image
- Tree style sets the shape, width and color of the clustering tree
- The "Data point shape" parameter sets the punctuation shape of the highest point of the data. There are 23 kinds of point shapes.
- "Color map" contains 166 types, mainly divided into the following four categories: Sequential colormaps: continuous colormaps; Diverging colormaps: colormaps with divergent ends; Qualitative colormaps: Miscellaneous colormaps: Other colormaps. Such as 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. For each suffix _r, the color graph is the original color bar flipped horizontally.
- Width and height parameters have been set
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 2024.8.18
Update time: 2024.8.18
Box plot
Use box plots to display data distribution and statistical summaries (e.g., median, quartiles) to help identify changes in gene expression or metabolic levels.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Set the X-axis and Y-axis on and off, font size, color, rotation angle and step length
- Set the label name and font size of the X-axis, Y-axis and label
- "Image resolution" parameter sets the image resolution
- " Add category column "for classification
- " Notch " sets whether to have a notch
- " Fill " sets whether the color is filled
- The "Transparency" parameter can adjust the transparency of the point color
- Set the legend title and title font size, legend font size, and legend position
- "Color map" contains 166 types, mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Set the legend title and title font size, legend font size, and legend position
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Multivariable histogram
Multivariable histograms are often used in biology to plot data of multiple variables or features to study their relationships and distributions.
Detailed instructions:
1. First click the file button on the right and select the Csv format or Excel format file that needs to be visualized
2. The default parameters have been set. If you need to adjust the parameters, please see below
Detailed instructions:
- Set the X-axis and Y-axis on and off, font size, color, rotation angle and step length
- Color bar sets its direction, label font size, label name, size, color, position, and font size.
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
3.After completing all configurations, click the "Run" button to display or update the heat map, and the heat map will appear in the canvas on the left.
4. Users can export the heat map by right-clicking the "Save" export option. hbiCloud provides 4 levels of images from 300 to 1000dpi for users to use. There are four image formats: png, .jpg, pdf, and .svg to meet different needs.
Online time: 2023.12 . 25
Update time: 2023.12 . 25
Bar chart
Use bar charts to display the frequency or mean of categorical data, which is suitable for comparing omics data such as gene expression and protein abundance.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Set the X-axis and Y-axis on and off, font size, color, rotation angle and step length
- Set the label name and font size of the X-axis, Y-axis and label
- "Image resolution" parameter sets the image resolution
- " Dot shape " sets 23 dot shapes
- " Transparency " parameter can adjust the transparency of the point color
- Set the legend title and title font size, legend font size, and legend position
- "Color map" contains 166 types, mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 3. 7
Updated: 2024. 3. 7
Line graph
Use line graphs to show changes in data over time or other continuous variables, which is suitable for the analysis of time series omics data.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Set the X-axis and Y-axis on and off, font size, color, rotation angle and step length
- Set the label name and font size of the X-axis, Y-axis and label
- "Image resolution" parameter sets the image resolution
- The " Line width " parameter sets the line width
- " Transparency " parameter can adjust the transparency of the point color
- Set the legend title and title font size, legend font size, and legend position
- "Color map" contains 166 types, mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 3. 7
Updated: 2024. 3. 7
Pie chart
Pie charts are used to display the proportion of classification data. They are suitable for displaying the composition of omics data such as gene classification and functional annotation.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- " Inner label unit " parameter sets the label format
- " Lable size " parameter sets the internal label font size
- There are 166 color maps, which are mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- "Image resolution" parameter sets the image resolution
- " Lable color " parameter label color
- " Line style " parameter border line style
- " Transparency " parameter sets the transparency " Startangle " parameter
- " pctdistance " parameter sets the ratio of the distance between the center of each pie slice and the beginning of the text generated by autopct to the radius
- " Startangle " parameter specifies the starting angle of the pie chart.
- " Label distance " parameter The radial distance at which pie chart labels are drawn
- " Radius " parameter: the radius of the pie chart
- The " Shadow " parameter determines whether to add a shadow
- " Counter clock " parameter whether to draw the fan counterclockwise
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 3. 7
Updated: 2024. 3. 7
Beeswarm plot
Similar to swarm plot, it better shows the distribution and density of data points and is suitable for detailed analysis of small-scale data.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Set the X-axis and Y-axis on and off, font size, color, rotation angle and step length
- Set the label name and font size of the X-axis, Y-axis and label
- "Image resolution" parameter sets the image resolution
- " Dot shape " sets 23 dot shapes
- " Box border color " sets the box border color
- " Dot size magnification " sets the magnification of the point
- for classificationSet the legend title and title font size, legend font size, and legend position
- "Color map" contains 166 types, mainly divided into the following four categories: Sequential colormaps: continuous colormaps, Diverging colormaps: diverging colormaps, Qualitative colormaps: discrete colormaps, Miscellaneous colormaps: other colormaps. For example, 'Accent', 'Accent_r', 'Blues', 'Blues_r', 'BrBG', 'BrBG_r', etc. Each colormap with the suffix _r is a horizontal flip of the original color bar.
- Set the legend title and title font size, legend font size, and legend position
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 4. 16
Update time: 2024. 4.16
Ternary plot
In multi-omics research, the ternary plot is an important visualization tool that can show the proportional relationship between three variables. The value of each variable represents a vertex, and the position of the data point represents the relative proportion of the three variables. In multi-omics research, ternary plots are often used to display the combination of different omics data, such as the relative abundance relationship of genes, metabolites, and proteins. This visualization method can help researchers intuitively understand the coordinated changes of multiple biological molecules in different conditions or samples, and provides strong support for revealing the interactions and functions in complex biological systems.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- Set the label name, font size, and color of the three elements
- The "Transparency" parameter can adjust the transparency of the point color
- Set the size of high expression points and the size of low expression points
- " No sign color " parameter sets the color of points without special marks
- Set the legend title and title font size, legend font size
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 4. 16
Update time: 2024. 4.16
Sankey plot
Use Sankey diagrams to show data flow and proportional changes, which are suitable for visualizing metabolic pathways or gene expression changes.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. Input parameters. If you need to adjust the parameters, please see below.
- "Sinks right" parameter whether to start from the right
- "Node width" parameter sets the node width
- The "Font size" parameter sets the font size
- "Node padding" parameter adjusts the node margins
- Set the width and height parameters
3. After completing all configurations, click the "Run" button to start running.
4. Display the output results in the Result area
Launch time: 2024. 3. 7
Updated: 2024. 3. 7
Stack plot
The stackplot tool is useful in multi-omics for visualizing the cumulative distribution of different omics layers over time or other metrics. It helps researchers understand the dynamic changes and relative contributions of various molecular entities, revealing patterns and interactions in complex datasets.
Detailed instructions:
1. First, select the input file format, and then click the "Upload" button in the "Upload data" area to input the files that need to be processed.
2. Input parameters. If you need to adjust the parameters, please see below.
3. After completing all configurations, click the "Run" button to start running.
4. The output results are displayed in the Result area.
Online time: 2024.8.13
Update time: 2024.8.13
Qrcode
Generate QR codes for quick sharing or storage of multi-omics data links, facilitating data exchange and access.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.6. 24
Update time: 2024.6. 24
Bar code
Generate barcodes for sample management and data tracking, ensuring the consistency and traceability of samples and data in multi-omics studies.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.6. 24
Update time: 2024.6. 24
md5 code
MD5 (Message Digest Algorithm 5) is a widely used cryptographic hash function that generates a 128-bit hash value (usually represented as a 32-bit hexadecimal number). MD5 is often used for data integrity verification and password storage.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.6. 24
Update time: 2024.6. 24
Blockchain code
Use blockchain technology to record and verify multi-omics data, ensure data integrity and security, and improve the credibility of data sharing.
Detailed instructions:
1. First, click the Upload button in the Upload data area to enter the file to be processed or enter the relevant data in the input box.
2. After completing all configurations, click the "Run" button to start running.
3. The output results are displayed in the Result area.
Launch time: 2024.6. 24
Update time: 2024.6. 24
 National Key Laboratory for Tropical Crop Breeding, Sanya Institute of Breeding and Multiplication,
National Key Laboratory for Tropical Crop Breeding, Sanya Institute of Breeding and Multiplication,